ASTMH 2017, Nicholas Brazeau: “Plasmodium vivax whole genome sequencing to assess genetic relatedness of polyclonal relapses in Cambodia”
Collaborator(s): University of North Carolina System, United States
Countries: Cambodia
Published: 08/11/2017
In collaboration with ASTMH, Image Audiovisuals, and session presenters, MESA brings you this webcast from the 66th ASTMH annual meeting in Baltimore, November 2017
Title: “Plasmodium vivax whole genome sequencing to assess genetic relatedness of polyclonal relapses in Cambodia”
Speaker: Nicholas Brazeau, University of North Carolina
Session information:
Symposium 0137: “Malaria: Genetics and Genomics”
Wednesday, 8 November, 10:15 – 12:00 PM, Convention Center – Room 321/322/323 (Level 300)
Abstract:
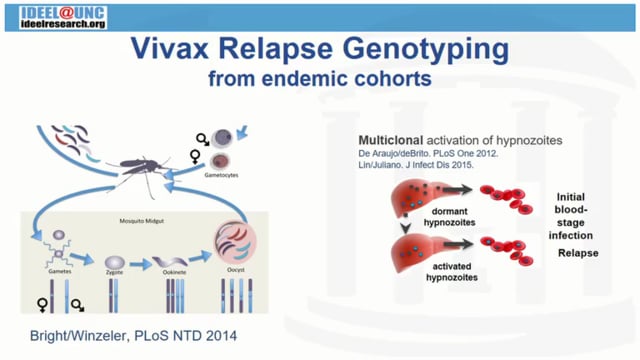
We previously performed targeted deep sequencing of pvmsp1 in a Cambodian cohort, including 22 persons who developed 29 Plasmodium vivax recurrences in the absence of primaquine treatment. We found frequent multiclonal infection and recurrences with homologous strains suggestive of relapse. We are now pursuing whole genome sequencing of these relapses. For 10 isolates from 3 patients who each suffered multiple relapses containing homologous strains, P. vivax DNA was enriched by hybrid capture, sheared, then sequenced on an Illumina HiSeq2000. Reads were aligned to the Sal-1 reference genome and variants called using the GATK pipeline. Coverage ≥5x was achieved across half the genome in 8/10 relapse isolates. We assessed genetic complexity and relatedness of the isolates in the context of 70 previously sequenced vivax genomes from the same region in northwestern Cambodia. The high complexity of infection found by deep sequencing was recapitulated on a genome-wide scale. 102,835 biallelic SNPs with 5x coverage across 80% of the 80 samples were used for an estimate of clonality based on the FWS statistic. All 10 relapse isolates were identified as polyclonal, with a median FWS of 0.60 (range 0.16-0.90). The FWS for each isolate decreased with successive episodes in 2/3 persons. Principal coordinates analysis of the 10 relapse genomes showed clustering by patient, indicating greater genetic relatedness among episodes within than between individuals. This genetic relatedness was recapitulated by STRUCTURE, which assigned isolates to three distinct “populations” corresponding to the three patients. Identity by descent analyses, with a framework to handle multiclonal infections, indicated a high degree of haplotype sharing among relapse samples within persons, consistent with our previous findings. All ten relapse samples aligned to the pvcsp VK210 clade. These analyses suggest that waves of reactivated hypnozoites within individuals are polyclonal and genetically related, with implications for distinguishing relapses from re-infections. Ongoing efforts are using whole genome amplification to sequence more relapses from the cohort.
Published: 08/11/2017
THEMES: Basic Science | P. vivax
Related Resources


